
Аномалия петерса помутнение роговицы
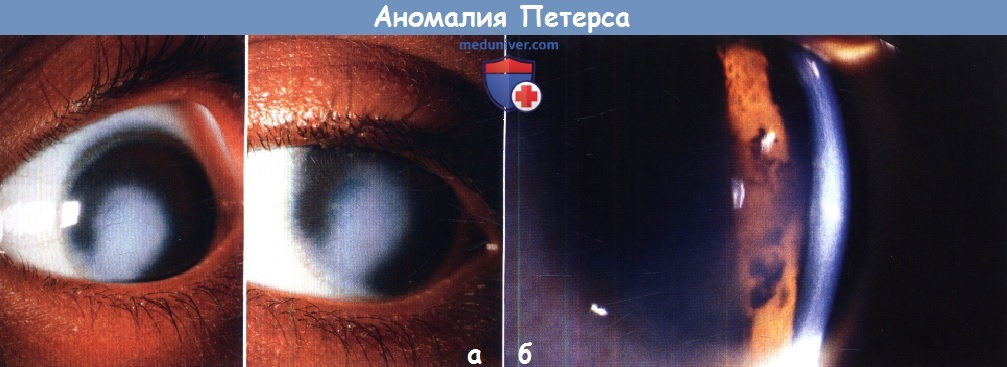
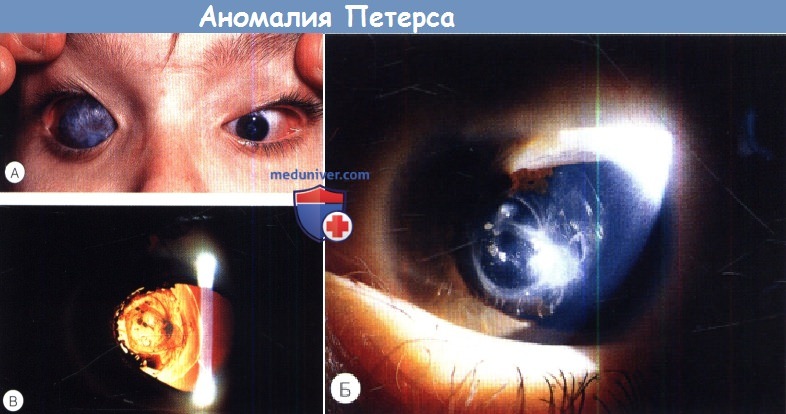
Аномалия Петерса у ребенкаПод названием аномалия Петерса обозначают иридокорнеальные или кератолентикулярные адгезии, приводящие к центральному или эксцентрическому, локальному или тотальному помутнению роговицы. Патогенез аномалии Петерса вызывает споры. Ни одна из теорий полностью не объясняет клинических и гистологических изменений при всех формах аномалии Петерса. Аномалию Петерса следует считать гетерогенной группой врожденных аномалий с одинаковыми клиническими проявлениями, помутнением роговицы, развивающихся в результате различных патогенных механизмов, в том числе генетических и/или под действием факторов среды. Возможно, в тех случаях, когда заболевание вызвано генетическими причинами, сопутствующие глазные нарушения могут указать на вызвавшую их мутацию; например, аниридия и аномалия Петерса, вероятно, развились вследствие мутации РАХ6, тогда как причиной аномалии/синдрома Аксенфельда-Ригера, видимо, являются изменения PITX2. Большинство случаев спорадические, но описано аутосомно-рецессивное и доминантное наследование. Аномалия Петерса определяется как врожденное центральное помутнение роговицы, сопровождающееся дефектами задних отделов стромы, десцеметовой мембраны и эндотелия. В 80% случаев поражение двустороннее. Глаукома развивается в 50-70% случаев. Аномалия Петерса подразделяется на три группы:
Самой простой и хуже всего описанной в литературе является форма аномалии Петерса, включающая в себя изолированный дефект задних слоев роговицы и бельмо. Радужка и хрусталик не изменены, но вследствие дефекта задних слоев роговицы развивается обширное помутнение, по плотности варьирующее от легкого облаковидного до выступающего над поверхностью васкуляризованного бельма, которое может уменьшаться в первые несколько лет жизни. Иногда дефект настолько выражен, что в центральной зоне возникает относительное просветление, окруженное мутной средней периферией роговицы. Периферия роговицы обычно прозрачна, что позволяет при помощи гониоскопа визуализировать роговично-хрусталиковые сращения, хотя часто наблюдается склерализация лимба. В недавней публикации мы описали мутации CYP1В1, связанные с врожденным помутнением роговицы и предположили, что соответствующая оригинальному описанию язва von Hippel может вызываться такой CYP1B1-«цитопатией»; за исключением глаукомы других глазных изменений не наблюдалось. Иридокорнеальные сращения обычно тянутся от брыжи радужки и варьируют от тонких нитей до широких тяжей. Могут развиваться кератолентикулярные сращения, которые бывают четырех различных типов: Сопутствующие глазные нарушения включают в себя синдром Аксенфельда-Ригера или аниридию, микрофтальмию, персистирующее гиперпластическое первичное стекловидное тело (persistent hyperplastic primary vitreous — PHPV) и дисплазию сетчатки. Сопутствующая системная патология включает в себя краниофациальные аномалии, врожденные пороки сердца, гипоплазию легких, синдактилию, аномалии уха, патологию мочеполовой системы, аномалии центральной нервной системы, карликовость, алкогольный синдром плода и хромосомные аномалии. Синдром Петерса-плюс — редкое аутосомно-рецессивное заболевание, проявляющееся карликовостью с короткими конечностями, сглаженным носогубным желобком и тонкой верхней губой, снижением слуха, расщелиной губы/неба, брахиморфизмом с короткими верхними конечностями, брахидактилией и сужающимися к концам пальцами, умственной отсталостью и двусторонней аномалией Петерса. Он связан с мутациями гена B3GALTL. Гистологические изменения могут варьировать, но они включают в себя утолщение боуменовой мембраны, легкие атрофические изменения вышележащего эпителия, нормальные передние слои стромы, сморщенные задние слои стромы, частично замещенные фиброзной тканью и обширный центральный дефект десцеметовой оболочки и эндотлия. На периферии роговицы обычно сохраняются интактные десцеметова мембрана и эндотелий. Передняя камера обычно глубокая, за исключением зон иридокорнеальных или кератолентикулярных сращений. Также описаны случаи отсутствия боуменовой мембраны с отеком передних и дефектом задних слоев стромы. Лечение врожденных помутнений роговицы исключительно трудно, и, несмотря на раннюю диагностику и рано начинаемое медикаментозное или хирургическое лечение, исход часто бывает неблагоприятным. Ранняя сквозная кератопластика в течение первых трех месяцев жизни дает младенцу наивысшие шансы на хорошее зрение. Швы снимают на 4-6 неделе, после чего подбирается контактная линза и проводится лечение амблиопии, которое бывает успешным только при условии сознательности и мотивированности всех участников. Хороший прием при консультации родителей — постоянно напоминать о цели лечения — достижении зрительных функций, соответствующих хотя бы нижней границе нормы. Однако отторжение трансплантата является серьезной проблемой, а данных о применении у младенцев искусственной роговицы, например, Boston К-pro, очевидно, все еще недостаточно. Однако, у детей, ранее перенесших кератопластику с последующим отторжением трансплантата, возможно применение Boston-К-pro. Альтернативы сквозной пластике аллотрансплантатом включают в себя обширную иридэктомию и ротационную аутокератопластику.
— Также рекомендуем «Склерокорнеа у ребенка» Оглавление темы «Аномалии переднего сегмента глаза у детей.»:
|
Источник
Ариткулова И.В.
Аномалия Петерса — редко встречающаяся врожденная патология переднего отдела глаза у детей с характерным патологическим симптомокомплексом дисгенеза элементов глазного яблока. Впервые заболевание описано А. Peters (18621938) в 1906 г. «Классическая» форма аномалии: врожденное центральное стромальное помутнение роговицы округлой формы и небольших размеров (2-3 мм) с истончением ее в зоне помутнения и иридокорнеальными сращениями — тяжами из ткани радужки, идущими от зрачкового края к кольцевидному фиброзному утолщению, по границе помутнения роговицы» [7].
Частота встречаемости заболевания в популяции 1:200 000. Приблизительно 700 чел. в России, 1500 чел. в СНГ и 35000 чел. во всем мире имеют аномалию Петерса [6].
Различают мезодермальную, эктодермальную и воспалительную формы заболевания. Первая обусловлена неполной абсорбцией или расщеплением мезодермы, связанной с центральной зоной роговицы и радужки. Десцеметова мембрана и эндотелий обычно отсутствуют в области помутнения, хрусталик остается интактным. При наличии передней полярной или субкапсулярной катаракты, вследствие неправильного отделения хрусталикового пузырька от поверхности эктодермы, ставится диагноз эктодермальной формы. Воспалительная форма, не являясь наследственной, развивается изза внутриутробного воспалительного процесса и может иметь признаки мезодермальной или эктодермальной формы.
Аномалия Петерса может сочетаться с аниридией, микрофтальмом, хориоретинальной колобомой, персистирующим первичным гиперпластическим стекловидным телом, врожденной афакией.
Описан «Петерс-плюс синдром», включающий помимо глазной симптоматики и ряд других аномалий, среди которых — волчья пасть, укорочение туловища и конечностей, задержка психомоторного развития. Кроме того, эта патология может входить в состав синдрома Краузе-Кивлина, проявляющегося помимо глазной симптоматики краниофациальным дисморфизмом, короткими конечностями, нарушением слуха. Около трети детей с аномалией Петерса имеют системную патологию (врожденные пороки сердца, патологию мочеполовой системы и др.) [5].
Аномалия Петерса наследуется по аутосомно-доминантному типу, хотя описан рецессивный тип наследования и спорадические случаи. У ряда больных обнаружены не являющиеся специфическими мутации в гене РАХ6, а также в генах FOXCI и PITX2 [8, 9].
На тактику лечения оказывает влияние выявление в половине случаев глаукомы, связанной с грубой врожденной деформацией угла передней камеры и иридокорнеальными сращениями и имеющей неблагоприятный прогноз для зрения.
Различают два клинических варианта заболевания [1]:
• Синдром Петерса типа I — типичное нубекулярное (облачковидное) центральное помутнение роговицы, окаймлённое тяжами радужки, которые пересекают переднюю камеру от зрачкового пояса радужки до роговицы, хрусталик прозрачный с правильным расположением. Глаукома присоединяется в 30% случаев.
• Синдром Петерса типа II — тяжёлое течение, имеется сращение хрусталика с центральной корнеальной лейкомой, выявляется передняя полярная катаракта. Часто ассоциируется с микрокорнеа, микрофтальмом, плоской роговицей, склерокорнеа, колобомой, аниридией и дисгенезом угла передней камеры и радужки. Глаукома развивается в 70% случаев.
Диагностика основана на данных соматического и офтальмологического обследования.
Дифференциальная диагностика: врождённая декомпенсированная глаукома, сопровождаемая помутнением роговицы и врождённым увеитом, кератитом.
Рекомендуется консервативное (местная тканевая терапия) и хирургическое лечение при выявлении высокого офтальмотонуса (синусотрабекулэктомия, оптическая иридотомия, частичная сквозная кератопластика, ленсэктомия с восстановлением передней камеры). В последние годы применяется лазерное рассечение иридокорнеальных сращений [2-4].
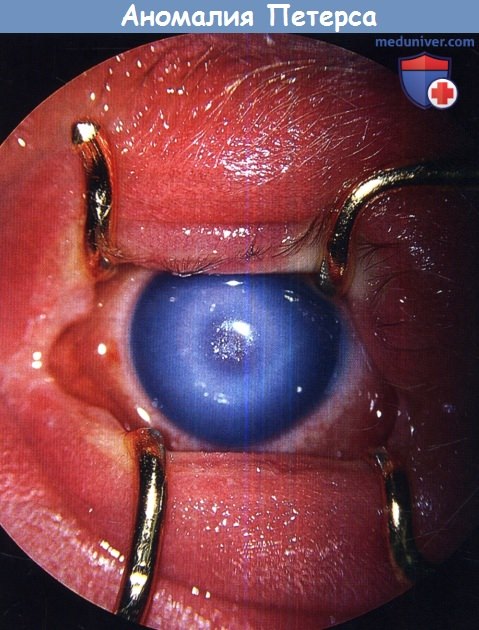
Представляем клинический случай аномалии Петерса у девочки 6 лет.
Ребёнок Б., 2007 г.р., наблюдается в УфНИИ ГБ с 3-месячного возраста с диагнозом врождённого центрального помутнения роговицы, микрокорнеа, иридокорнеальных сращений, частичного помутнения хрусталика, вторичного расходящегося косоглазия с вертикальным компонентом (последствия внутриутробного кератоувеита) левого глаза; гиперметропией слабой степени, сложного гиперметропического астигматизма правого глаза.
Из анамнеза: мать ребёнка с 1995 по 2006 гг. наблюдалась и лечилась у кожвенеролога вследствие положительной реакции Вассермана, с учёта снята.
У ребёнка антитела в крови к сифилису не обнаружены. Ребёнок от III беременности, роды I, преждевременные, в срок 8 мес. кесарево сечение в связи со стойким повышением артериального давления у матери. Диагноз при рождении: гипоксически-ишемическое поражение центральной нервной системы. Наблюдается с диагнозом резидуальной энцефалопатии, синдромом внутричерепной гипертензии у невролога. В анамнезе острая пневмония и частые ОРВИ.
Предметное зрение OS с рождения отсутствует (помутнение роговицы в оптической зоне). OS: микрокорнеа, глазное яблоко отклонено кнаружи до 10°, книзу до 3°, движения в полном объёме, конвергенция ослаблена, конъюнктива бледно-розовая, в оптической зоне роговицы помутнение до 4 мм в диаметре, более интенсивное в центре, по периферии — пигмент на эндотелии, передняя камера в центре отсутствует, по периферии — мелкая, неравномерная, в верхнем секторе виден внутренний край зрачка неправильной формы, ригидный, выражены крипты радужки, иридокорнеальные сращения — тяжи из ткани радужки, идущие от зрачкового края к кольцевидному утолщению на границе помутнения роговицы, хрусталик и стекловидное тело не просматриваются, рефлекса с глазного дна нет. OD — без особенностей.
Острота зрения: правый глаз — 0,9, с коррекцией cyl +0,5 D ax 100° — 0,9-1,0; OS — 1/? pr. сrt.
Внутриглазное давление пальпаторно — нормотонус, бесконтактным методом 23,0/35,0 мм рт.ст., по Шиотцу 20,6/22,4 мм рт.ст. — данные недостоверны, так как имеем следующие результаты пахиметрии: ОD — 561-571-587 мм (центр), OS — 784-921-990 мм (центр), что свидетельствует о значительной плотности роговицы в центре, особенно на левом глазу.
Горизонтальный диаметр роговицы: ОD — 11,5 мм, OS — 10,0 мм. Гониоскопия: OS — передние периферические синехии, УПК частично закрыт. Ультразвуковая биометрия: ОD — 21,83 мм; 3,11 мм; 3,49 мм; ОS — 25,63 мм; 2,08 мм; 3,10 мм.
Данные авторефрактометрии: ОD sph +1,5 D cyl +0,75 D ax 100°; OS — не определяются. В-сканирование: OS — на ультрасонограмме в стекловидном теле акустически гетерогенные включения в виде точек, не фиксированных к сетчатой оболочке, отслойки оболочек глаза нет, ретробульбарная область без патологии, канал зрительного нерва нормальный.
Ребенок получал консервативное лечение: электрофорез с лидазой, витаминотерапию, антиоксиданты, сосудорасширяющие препараты. В последующем планируется проведение на левом глазу сквозной частичной кератопластики с оптической целью.
Выводы. Проведенный анализ клинических проявлений и тактики ведения больных с редкой врождённой аномалией глаза у детей — аномалией Петерса — свидетельствует о необходимости дифференциальной диагностики данного заболевания с последствиями воспалительного процесса в глазу: врождённым увеитом и кератитом, врождённой декомпенсированной глаукомой, для правильной оценки тяжести заболевания и выбора метода лечения.
Источник
УДК 617.713-004.1
А.В. ТЕРЕЩЕНКО, И.Г. ТРИФАНЕНКОВА, М.С. ТЕРЕЩЕНКОВА
Калужский филиал «МНТК «Микрохирургия глаза» им. акад. С.Н. Федорова» МЗ РФ, 248007, г. Калуга, ул. Св. Федорова, д. 5
Терещенко Александр Владимирович ― доктор медицинских наук, директор, тел. (4842) 505-767, e-mail: [email protected]
Трифаненкова Ирина Георгиевна ― кандидат медицинских наук, заместитель директора по научной работе, тел. (4842) 505-767, e-mail: [email protected]
Терещенкова Маргарита Сергеевна ― кандидат медицинских наук, заведующая детским хирургическим отделением, тел. (4842) 505-767, e-mail: [email protected]
В работе рассмотрены вопросы патогенеза, диагностики и лечения тяжелой врожденной патологии ― аномалии Петерса. По мнению специалистов, точная причина возникновения данного заболевания не установлена, однако, многие исследователи указывают на ее генетическую этиологию. Согласно данным литературы, на сегодняшний день существует целый ряд различных хирургических вариантов лечения аномалии Петерса. Для определения оптимальной тактики и сроков проведения хирургического вмешательства при данной патологии важным является тщательное диагностическое обследование ребенка.
Ключевые слова: врожденная патология, аномалия Петерса, пересадка роговицы.
A.V. TERESHCHENKO, I.G. TRIFANENKOVA, M.S. TERESHCHENKOVA
Kaluga branch of the «Interbranch Scientific and Technical Complex «Eye Microsurgery» named after the acad. S. N. Fedorov» of the Ministry of Health of the Russian Federation, 5 Sv. Fedorova Str., Kaluga, Russian Federation, 248007
Peter’s anomaly
Tereshchenko A.V. ― D. Med. Sc., Director, tel. (4842) 505-767, e-mail: [email protected]
Trifanenkova I.G. ― Cand. Med. Sc., Deputy Director for Science, tel. (4842) 505-767, e-mail: [email protected]
Tereshchenkova M.S. ― Cand. Med. Sc., Head of Children’s Surgical Department, tel. (4842) 505-767, e-mail: [email protected]
This article considers the issues of pathogenesis, diagnosis and treatment of severe congenital abnormality ― Peter’s anomaly. Experts say that the exact cause of this disease has not been established, however, many researchers point to its genetic etiology. According to literature, at the moment there are a number of different surgical options for the treatment of Peter’s anomaly. To determine the optimal tactics and timing of surgery in case of this disease, it is important to make a thorough diagnostic examination of a child.
Key words: congenital abnormality, Peter’s anomaly, corneal transplantation.
Аномалия Петерса ― достаточно редко встречающийся врожденный дисгенез переднего отрезка глаза у детей. В 1906 году Альфред Петерс впервые описал связь между дефектом десцеметовой мембраны, неравномерной передней камерой, иридокорнеальными синехиями и помутнением роговицы (лейкомой). Позже для описания данного синдрома стали использовать термин «аномалия Петерса». Спектр состояний, обозначенных как аномалия Петерса, на сегодняшний день расширен и может включать случаи одностороннего или двустороннего поражения глаз, с системными поражениями или без них.
Частота заболевания в популяции составляет 1:200000. Согласно данным литературы, аномалия Петерса зафиксирована приблизительно в 700 случаях в России, а во всем мире около 35000 человек имеют разные формы данной патологии.
В 1974 году W. Townsend с соавторами предложили классификацию аномалии Петерса, где были выделены 3 основных типа: 1 ― изолированное центральное помутнение роговицы (лейкома), 2 ― центральное помутнение роговицы с корнеолентикулярным сращением, 3 ― центральное помутнение роговицы с мезодермальным дисгенезом Ригера [1]. Однако, в дальнейшем, вариации данных классификационных категорий были пересмотрены и определены следующим образом: тип 1 (мезодермальный) ― врожденное центральное стромальное помутнение роговицы чаще округлой формы с истончением ее в зоне помутнения и иридокорнеальными сращениями; тип 2 (эктодермальный) ― центральное помутнение роговицы и иридокорнеолентикулярные сращения разной степени выраженности, зачастую возможно не только помутнение хрусталика, но и смещение его кпереди, нередко с отсутствием его дифференцировки и как бы сращением с задней поверхностью роговицы, передняя камера мелкая, неравномерная, местами отсутствует; тип Петерс-плюс ― симптомокомплекс глазных проявлений аномалии Петерса, который ассоциируется также с другой глазной патологией (глаукома, микрофтальм, хориоретинальные колобомы, перфорации роговицы, аниридия, персистирующее первичное гиперпластическое стекловидное тело, врожденная афакия) и системными поражениями, такими как врожденные пороки сердца или почек, половых органов, аномалии центральной нервной системы (агенезия мозолистого тела, задержка развития, внутричерепные кальцификаты), челюстно-лицевые дефекты (заячья губа, волчья пасть, микрогнатия, зубные дефекты, низко посаженные уши), дефекты опорно-двигательного аппарата (брахидактилия, клинодактилия, короткие конечности, низкорослость, позвоночные аномалии). В 60-80% случаев процесс носит двусторонний характер [2-5].
К настоящему времени не удалось выяснить точную причину аномалии Петерса. Тем не менее, многие авторы указывают на ее генетическую этиологию.
В обзорной статье, опубликованной в 2011 году, Bhandari и коллеги пришли к выводу, что аномалия Петерса зачастую возникает спорадически [3]. Однако в нескольких семьях был зарегистрирован как аутосомно-доминантный, так и аутосомно-рецессивный характер передачи заболевания [6]. Описаны ряд хромосомных аномалий и специфические генетические мутации, которые были связаны с аномалией Петерса. У части больных обнаружены мутации в гене РАХ6 и REIG1, а также в генах PITX2 и FOXE3 [7-9].
В своей статье M. Takamiya и соавт. пришли к выводу, что человеческие PAX6 мутации связаны с некоторыми глазными заболеваниями, одним из которых является аномалия Петерса. Кроме того, генетические мутации в бета-1,3-галактозилтрансферазе гена B3GALTL могут вызвать синдром Петерс-плюс, в то время как микроделеции в 8q21.11 хромосоме ― непостоянное проявление аномалии Петерса [10-12].
Возникновение аномалии Петерса происходит на 4-7 неделе эмбрионального развития в результате нарушения отделения хрусталикового пузырька от поверхности эктодермы или же неполной абсорбции и расщепления мезодермы, связанной с центральной и парацентральной зонами радужки и роговицы во время развития передней камеры. При гистологическом исследовании обнаруживается отсутствие десцеметовой мембраны и эндотелия в участке помутнения роговицы [13].
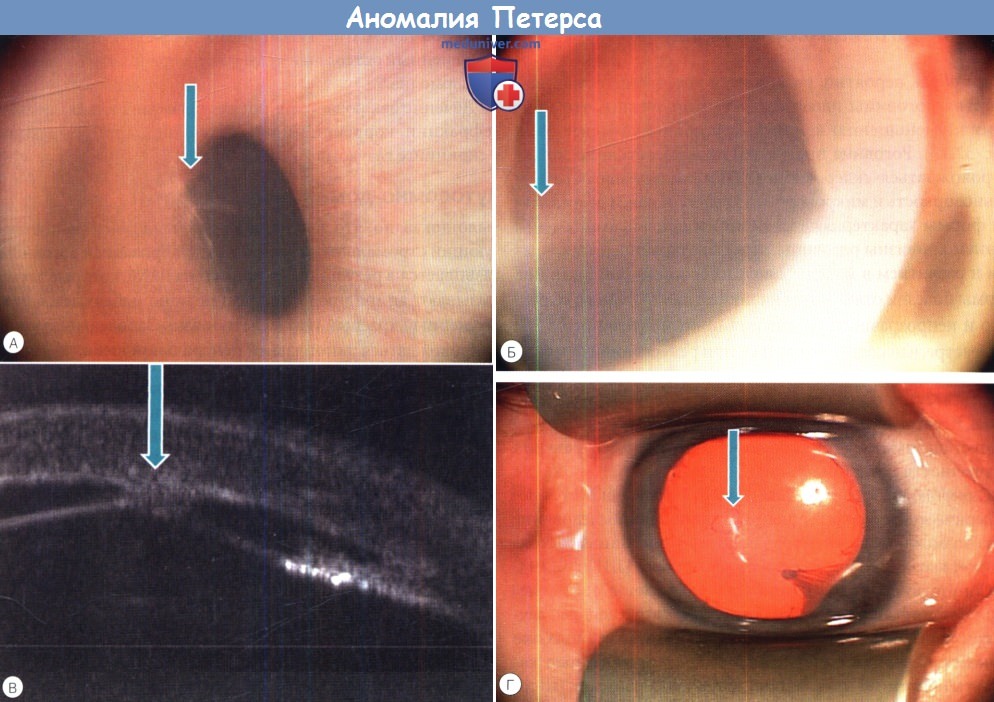
Важным этапом в определении оптимальной тактики и сроков проведения хирургического вмешательства при аномалии Петерса является тщательное диагностическое обследование ребенка. Помимо общепринятых методов в последние годы у детей активно и широко применяются оптическая когерентная томография (ОСТ) и ультразвуковая биомикроскопия (УБМ). Это высоко информативные и объективные методы визуализации переднего отрезка глаза, позволяющие получить и интерпретировать изображения структур роговицы, передней камеры, радужки, хрусталика, цилиарного тела и его отростков, связочного аппарата хрусталика и их соотношение. Исследователи указывают на высокую информативность данных методов не только для определения типа аномалии, но и степени ее выраженности [14, 15].
Лечебные мероприятия при аномалии Петерса направлены на своевременное выявление и лечение сопутствующей глаукомы, а также, по возможности, обеспечение прозрачности хрусталика и оптического центра роговицы в зависимости от клинического типа течения. На сегодняшний день существует целый ряд различных хирургических вариантов, доступных для лечения аномалии Петерса. Для предотвращения развития обскурационной амблиопии лечение должно проводиться в младшем возрасте.
Как уже отмечалось, одним из симптомов аномалии Петерса является глаукома, которая развивается в 20-50% случаев как следствие аномального строения угла передней камеры и оказывает существенное влияние на тактику лечения и на дальнейшее развитие зрительных функций. Для лечения офтальмогипертензии применяются следующие виды хирургических вмешательств: синустрабекулэктомия, иридоциклоретракция или деструктивные вмешательства на цилиарном теле как методы стойкого гипотензивного эффекта. После нормализации офтальмотонуса становится возможным проведение дальнейшего лечения.
Следующим важным этапом в лечении аномалии Петерса является восстановление прозрачности оптических сред: роговицы и/или хрусталика. В ряде случаев при помутнениях роговицы и хрусталика небольшой площади и малой интенсивности у детей младшего возраста отдается предпочтение местной рассасывающей и тканевой терапии с последующим щадящим вариантом оперативного лечения ― оптической (секторальной, периферической) иридэктомии. Суть подобной операции сводится к расширению зрачка до диаметра, превышающего размеры лейкомы [5]. Кроме того, в лечении аномалии Петерса традиционно используются такие хирургические методы как реконструкция передней камеры с хирургическим рассечением передних сращений, при необходимости в сочетании с факоаспирацией врожденной катаракты, а в последнее время ― факоаспирацией с фемтосекундным сопровождением [16-18].
Рассечение передних сращений в передней камере производят не только операционным доступом, но и с помощью лазера непосредственно у места сращения радужки с роговицей, с фокусировкой излучения непосредственно у места фиксации сращения радужки с участком мутной роговицы.
Преобладающее число работ при аномалии Петерса посвящено применению пересадки роговицы. Лечение синдрома заключается не только в проведении сквозной кератопластики с реконструкцией передней камеры, но и в комбинированной операции при 2 типе — с удалением хрусталика (возможны разные варианты: ленсэктомия, факоаспирация с имплантацией ИОЛ). Однако пересадка роговицы в раннем детском возрасте зачастую сопряжена с многочисленными техническими трудностями, кроме того, затруднен послеоперационный уход за данным контингентом пациентов, поэтому большинство авторов указывают, что проведение кератопластики целесообразно лишь в случаях тяжелого двустороннего поражения роговицы [19, 20].
По данным литературы, сквозная кератопластика при аномалии Петерса в обязательном порядке проводилась в случаях спонтанной перфорации роговицы с органосохранной целью. На сегодняшний день опубликованы только три сообщения о спонтанной перфорации роговицы при данном синдроме: U. Krause с соавторами сообщил о первом случае в 1969 году, E. Traboulsi и I. Maumenee ― в 1992 году, C. Banning с коллегами. описал двух пациентов с перфорацией роговицы и вторичной врожденной афакией при аномалии Петерса в 2005 году [21-23].
Сроки проведения сквозной кератопластики, по данным литературы, сильно варьируют ― от первого месяца после рождения до 7-12 месяцев. Процент прозрачного приживления трансплантата у разных авторов колеблется от 30 до 50%. Скорость отторжения трансплантанта в течение 1 года после операции находится в довольно широком диапазоне, от 22 до 67%. Довольно большое (до 60%) число повторных операций связано с недостаточной прозрачностью трансплантата [24-27].
Таким образом, аномалия Петерса представляет собой серьезную врожденную патологию, требующую своевременного проведения комплексного диагностического обследования, выбора оптимальной тактики с применением современных технологий хирургического лечения.
ЛИТЕРАТУРА
- Townsend W., Font R.L., Zimmerman L. Congenital corneal leukoma. Histopathological findings in 19 eyes with central corneal defects in Descemet’s membrane // Am. J. Ophthalmol. ― ― №77. ― P. 192.
- Zaidman G.W., Flanigan J.K., Furey C.C. Long-term visual prognosis in children after corneal transplant surgery for Peters anomaly type I // Am. J. Ophthalmol. ― ― №144. ― P. 104-108.
- Bhandari R., Ferri S., Whittaker B., et al. Peters anomaly: review of the literature // Cornea. ― ― №30. ― P. 939-944.
- Najjar D.M., Christiansen S.P., Bothun E.D., Summers C.G. Strabismus and amblyopia in bilateral peters anomaly // JAAPOS. ― ― №10. ― P. 193-197.
- Боброва Н.Ф., Тронина С.А. Особенности хирургического и консервативного лечения аномалии развития глаза (аномалии петерса) у детей // Офтальмологический журнал. ― ― №4. ― С. 20-24.
- Frydman M., Weinstock A.L., Cohen H.A., et al. Autosomal recessive Peters anomaly, typical facial appearance, failure to thrive, hydrocephalus, and other anomalies: further delineation of the Krause-Kivlin syndrome // Am. J. Med. Genet. ― ― №40. ― P. 34-40.
- Dahl E., Koseki H., Balling R. Pax genes and organogenesis // Ophthalmic Surg Lasers. ― ― №28. ― P. 311-312.
- Doward W., Perveen R., Lloyd I.C., et al. A mutation in REIG1 gene associated with Peters’ anomaly // J. Med. Genet. ― ― №36. ― P. 152-155.
- Iseru S.U., Osbourne R.J., Farrall M., et al. Seeing clearly: the dominant and recessive nature of FOXE3 in eye developmental anomalies // Hum Mutat. ― ― №10. ― P. 1378-1386.
- Takamiya M., Weger B.D., Schindler S., et al. Molecular description of eye defects in the zebrafish Pax6b mutant, sunrise, reveals a Pax6b-dependent genetic network in the developing anterior chamber // PLoS One. ― ― №10. ― P. 1176-1185.
- Denie K.F., Wesseling P., Eggink C.A. Unique presentation of corneal opacity in Peters plus syndrome: an unusual form of Peters anomaly showing tissue repair in serial analysis // Cornea. ― ― №35. ― P. 277-280.
- Happ H., Schilter K.F., Weh E., et al. 8q21.11 microdeletion in two patients with syndromic Peters anomaly // Am. J. Med. Genet A. ― ― №170. ― P. 2471-2475.
- Matsubara A., Ozeki H., Matsunaga N., et al. Histopathological examination of two cases of anterior staphyloma associated with Peters’ anomaly and persistent hyperplastic primary vitreous // Br. J. Ophthalmol. ― ― Vol. 85, №12. ― P. 1421-1425.
- Hong J., Yang Y., Cursiefen C., Mashaghi A., et al. Optimising keratoplasty for Peters’ anomaly in infants using spectral-domain optical coherence tomography // Br. J. Ophthalmol. ― ― №22. ― P. 658.
- Плескова А.В., Катаргина Л.А., Мазанова Е.В. Ультразвуковая биомикроскопия в диагностике врожденных помутнений роговицы у детей // Российская педиатрическая офтальмология. ― ― №1. ― С. 30-32.
- Ковалевский Е.И. Глазные болезни. Атлас. ― М.: Медицина, 1985.
- Tadayuki N., Misako N., Natsuki H. et al. Cataract Surgery for Tilted Lens in Peters’ Anomaly Type 2 // Case Rep Ophthalmol. ― ― Vol. 4, №3. ― P. 134-137.
- Hou J.H., Crispim J., Cortina M.S., Cruz Jde L. Image-guided femtosecond laser-assisted cataract surgery in Peters anomaly type 2 // J. Cataract Refract. Surg. ― ― Vol. 41, №11. ― P. 2353-7.
- Зубарева JI.H., Овчинникова А.В., Коробкова Г.В. Результаты сквозной кератопластики у детей // Офтальмохирургия. ― ― №3. ― С. 15-22.
- Слонимский А.Ю. Возможности сквозной пересадки роговицы при различной патологии переднего отрезка глаза // Клиническая офтальмология. ― ― Т. 2, №3. ― С. 21-26.
- Krause U., Koivisto M., Rantakallio P. A case of Peters syndrome with spontaneous corneal perforation // J. Pediatr Ophthalmol. Strabismus. ― ― №6. ― P. 145-149.
- Traboulsi E.I., Maumenee I.H. Peters’ anomaly and associated congenital malformations // Arch. Ophthalmol. ― ― №110. ― P. 1739-1742.
- Banning C.S., Blackmon D.M., Song C.D., Grossniklaus H.E. Corneal perforation with secondary congenital aphakia in Peters’ anomaly // Cornea. ― ― №24. ― P. 118-120.
- Chang J.W., Kim J.H., Kim S.J., Yu Y.S. Long-term clinical course and visual outcome associated with Peters’ anomaly // Eye (Lond). ― ― Vol. 26, №9. ― P. 1237-1242.
- Dana M.R., Schaumberg D.A., Moyes A.L., Gomes J.A. Corneal transplantation in children with peters anomaly and mesenchymal dysgenesis. multicenter pediatric keratoplasty study // Ophthalmology. ― ― №104. ― P. 1580-1586.
- Kim Y.W., Choi H.J., Kim M.K., et al. Clinical outcome of penetrating keratoplasty in patients 5 years or younger: peters anomaly versus sclerocornea // Cornea. ― ― №32. ― P. 1432-1436.
- Limaiem R., Chebil A., Baba A. Pediatric penetrating keratoplasty: indications and outcomes // Transplant Proc. ― ― №43. ― P. 649-651.
REFERENCES
- Townsend W., Font R.L., Zimmerman L. Congenital corneal leukoma. Histopathological findings in 19 eyes with central corneal defects in Descemet’s membrane. Am. J. Ophthalmol, 1974, no. 77, p. 192.
- Zaidman G.W., Flanigan J.K., Furey C.C. Long-term visual prognosis in children after corneal transplant surgery for Peters anomaly type I. Am. J. Ophthalmol, 2007, no. 144, pp. 104-108.
- Bhandari R., Ferri S., Whittaker B., et al. Peters anomaly: review of the literature. Cornea, 2011, no. 30, pp. 939-944.
- Najjar D.M., Christiansen S.P., Bothun E.D., Summers C.G. Strabismus and amblyopia in bilateral peters anomaly. JAAPOS, 2006, no. 10, pp. 193-197.
- Bobrova N.F., Tronina S.A. Features of surgical and conservative treatment of anomalies in the development of the eye (anomaly of the peters) in children. Oftal’mologicheskiy zhurnal, 2001, no. 4, pp. 20-24 (in Russ.).
- Frydman M., Weinstock A.L., Cohen H.A., et al. Autosomal recessive Peters anomaly, typical facial appearance, failure to thrive, hydrocephalus, and other anomalies: further delineation of the Krause-Kivlin syndrome. Am. J. Med. Genet, 1991, no. 40, pp. 34-40.
- Dahl E., Koseki H., Balling R. Pax genes and organogenesis. Ophthalmic Surg Lasers, 1997, no. 28, pp. 311-312.
- Doward W., Perveen R., Lloyd I.C., et al. A mutation in REIG1 gene associated with Peters’ anomaly. J. Med. Genet, 1999, no. 36, pp. 152-155.
- Iseru S.U., Osbourne R.J., Farrall M., et al. Seeing clearly: the dominant and recessive nature of FOXE3 in eye developmental anomalies. Hum Mutat, 2009, no. 10, pp. 1378-1386.
- Takamiya M., Weger B.D., Schindler S., et al. Molecular description of eye defects in the zebrafish Pax6b mutant, sunrise, reveals a Pax6b-dependent genetic network in the developing anterior chamber. PLoS One, 2015, no. 10, pp. 1176-1185.
- Denie K.F., Wesseling P., Eggink C.A. Unique presentation of corneal opacity in Peters plus syndrome: an unusual form of Peters anomaly showing tissue repair in serial analysis. Cornea, 2016, no. 35, pp. 277-280.
- Happ H., Schilter K.F., Weh E., et al. 8q21.11 microdeletion in two patients with syndromic Peters anomaly. Am. J. Med. Genet A, 2016, no. 170, pp. 2471-2475.
- Matsubara A., Ozeki H., Matsunaga N., et al. Histopathological examination of two cases of anterior staphyloma associated with Peters’ anomaly and persistent hyperplastic primary vitreous. Br. J. Ophthalmol, 2001, vol. 85, no. 12, pp. 1421-1425.
- Hong J., Yang Y., Cursiefen C., Mashaghi A., et al. Optimising keratoplasty for Peters’ anomaly in infants using spectral-domain optical coherence tomography. Br. J. Ophthalmol, 2016, no. 22, p. 658.
- Pleskova A.V., Katargina L.A., Mazanova E.V. Ultrasonic biomicroscopy in diagnosis of congenital opacities of the cornea in children. Rossiyskaya pediatricheskaya oftal’mologiya, 2014, no. 1, pp. 30-32 (in Russ.).
- Kovalevskiy E.I. Glaznye bolezni. Atlas [Eye diseases. Atlas]. Moscow: Meditsina, 1985.
- Tadayuki N., Misako N., Natsuki H. et al. Cataract Surgery for Tilted Lens in Peters’ Anomaly Type 2. Case Rep Ophthalmol, 2013, vol. 4, no. 3, pp. 134-137.
- Hou J.H., Crispim J., Cortina M.S., Cruz Jde L. Image-guided femtosecond laser-assisted cataract surgery in Peters anomaly type 2. J. Cataract Refract. Surg, 2015, vol. 41, no. 11, pp. 2353-7.
- Zubareva L.N., Ovchinnikova A.V., Korobkova G.V. Results of a through keratoplasty in children. Oftal’mokhirurgiya, 2000, no. 3, pp. 15-22 (in Russ.).
- Slonimskiy A.Yu. Opportunities of a through transplantation of a cornea at a various pathology of a forward segment of an eye. Klinicheskaya oftal’mologiya, 2001, vol. 2, no. 3, pp. 21-26 (in Russ.).
- Krause U., Koivisto M., Rantakallio P. A case of Peters syndrome with spontaneous corneal perforation. J. Pediatr Ophthalmol. Strabismus, 1969, no. 6, pp. 145-149.
- Traboulsi E.I., Maumenee I.H. Peters’ anomaly and associated congenital malformations. Arch. Ophthalmol, 1992, no. 110, pp. 1739-1742.
- Banning C.S., Blackmon D.M., Song C.D., Grossniklaus H.E. Corneal perforation with secondary congenital aphakia in Peters’ anomaly. Cornea, 2005, no. 24, pp. 118-120.
- Chang J.W., Kim J.H., Kim S.J., Yu Y.S. Long-term clinical course and visual outcome associated with Peters’ anomaly. Eye (Lond), 2012, vol. 26, no. 9, pp. 1237-1242.
- Dana M.R., Schaumberg D.A., Moyes A.L., Gomes J.A. Corneal transplantation in children with peters anomaly and mesenchymal dysgenesis. multicenter pediatric keratoplasty study. Ophthalmology, 1997, no. 104, pp. 1580-1586.
- Kim Y.W., Choi H.J., Kim M.K., et al. Clinical outcome of penetrating keratoplasty in patients 5 years or younger: peters anomaly versus sclerocornea. Cornea, 2013, no. 32, pp. 1432-1436.
- Limaiem R., Chebil A., Baba A. Pediatric penetrating keratoplasty: indications and outcomes. Transplant Proc, 2011, no. 43, pp. 649-651.
Источник

